

Pediatric mediastinal lymphoma
Introduction
Mediastinum, a visceral compartment of the thoracic cavity plays a niche in various organs of the human body. Based on the anatomy, the mediastinum is divided into superior and inferior mediastinum which is further subdivided into compartments. Anterior, middle, and posterior mediastinum from the inferior mediastinum. Anterior mediastinum mainly contains thymus and lymph nodes. The middle mediastinum contains heart, major vessels, and posterior mediastinum homes esophagus, and descending thoracic aorta (Figure 1). Lymph nodes are confined majorly to the anterior mediastinum; however, they can be found in any of the compartments. The intrathoracic lymph nodes are divided into fourteen sub-groups based upon their locations ascertained by mediastinoscopy. The 1–9 nodes correspond to mediastinal nodes, 10–14 are hilar and others include extra mediastinal like superior, inferior, and aortic nodes (1).

Thymus, one of the central lymphoid organs for the maturation of the T cells comprises the histological division of cortex and medulla. It predominantly comprises of immune cells, epithelial cells, and scant stromal cells. Epithelial cells form a complex network within which the T-cells, B-cells, histiocytes, and stromal cells are present (2). After traveling from the bone marrow, T-cells migrate to the thymic cortex and become subcapsular cortical thymocyte (stage I thymocyte) having an immature T-cell phenotype. They express terminal deoxynucleotidyl transferase (TdT), CD1a, CD3, CD5, and CD7 (Figure 2). From the initial double-negative phenotype for CD4 and CD8, they progressively mature into double-positive type with co-expression of the antigens and become stage II thymocyte or double-positive T cells. With a series of maturation steps in the thymic cortex and medulla, naïve T cells are formed which are either positive for CD4 or CD8 and are known as medullary thymocyte (stage III thymocyte). Medullary thymocytes have a phenotype similar to that of mature T-cells in the peripheral lymphoid organs (3).

In comparison to T-cell numbers, fewer B-cells are present in the thymus. Hofmann et al. coined the term asteroid B cells for the thymic B cells due to their cytoplasmic process. These cells are abundant in the cortico-medullary junctions and medulla (4). Two theories are postulated regarding the origin of thymic B-cell (5). These cells appear early in embryonic life before the origin of hematopoietic progenitor cells; hence are thought to have a development independent of marrow development (6). Few other authors postulate that the circulating multipotent hematopoietic stem cells and early precursor cells home in the thymus and further develop into a mature B cell (5). However, irrespective of the origin, T-cells are involved in the regulation of B-cell development proven by the expanded B-cell population in the mice model with disrupted T-cell pathway NOTCH signaling, a major pathway for T cell development also influences the thymic B cell population (7). The clear nature of these cells is not known; however, these resemble post germinal center B cells and are immunopositive for CD20, CD23, IgM while are negative for other dendritic cell markers like CD21 and CD35 (8,9). Primary mediastinal lymphoma is the lymphoma involving thymus, mediastinal lymph nodes, and mediastinal organs like heart, lung, pleura, and pericardium without any evidence of involvement of distant organs at the time of presentation (10). For all practical purposes, primary mediastinal lymphoma refers to the lymphoma arising from the central lymphoid organ, thymus. We will mainly focus on the lymphoma arising from the thymus and more common mediastinal lymphoma Hodgkin lymphoma.
The primary lymphomas of the mediastinum are: (Figure 3) (10).

Lymphomas comprise about 12% of all the mediastinal tumors and can be primary or part of systemic involvement (11). Few studies show lymphomas forming a huge burden (50%) of the pediatric mediastinal masses (12). Among the pediatric mediastinal lymphomas, lymphoblastic lymphoma (LBL) predominate followed by Hodgkin lymphoma (HL), primary mediastinal large B cell lymphoma (PMBCL) and very rarely Grey zone lymphoma. Tansel et al. reported an equal prevalence of Hodgkin and LBL among the pediatric population (13). Other types of aggressive and indolent non-HLs (NHLs) can occur in mediastinal lymph nodes and mediastinal organs (heart, lung, pleura, and pericardium) in adults; however, they are extremely rare in pediatric age group. Mediastinal lymphomas can be broadly divided based upon the cell of origin into the immature (blastic) type and mature. Though clinically and radiologically the anterior mediastinal mass has a similar presentation, accurate pathological diagnosis is essential for appropriate treatment (Figure 4). In this review, we will discuss the most common mediastinal lymphomas T-LBL, HL, PMBCL and their differential diagnosis (14-16).

Methods
This is a narrative review. A detail literature search was done for mediastinal lymphomas, pediatric mediastinal tumors published in PubMed and Google scholar in English literature between 2000–2020. For thymic B cell characterization older literature published between 1990–2000 were also searched. We present this article in accordance with the Narrative Review checklist (available at http://dx.doi.org/10.21037/med-20-37).
LBL
Introduction
LBLs are a group of aggressive neoplasms of the precursor immature cells of lymphoid lineage either committed to the T- or B-lineage. T-LBL manifests as nodal or extranodal masses with less than 25% blasts in the marrow; more than of which corresponds to leukemia. However, due to lack of consensus, WHO suggests the diagnosis of leukemia should be avoided if blast count <20% (17). As indicated by various studies, mediastinal involvement is a common occurrence in both the pediatric and adult populations. Thymus, the primary lymphoid organ for the maturation of T cells forms the maximally affected site for T-LBL. World health organization classification includes both T-LBL and acute lymphoblastic leukemia (ALL) under a common umbrella, precursor T-ALL/LBL, however, we will focus on T-LBL (17).
Incidence
LBL comprises approximately 2% of all the NHLs and constitutes 30% of Pediatric NHLs (18). T-LBLs predominate accounting for 85–90% of the LBLs whereas B-LBLs are a rare occurrence (18,19). However, B-cell ALL are commoner than their T-cell counterpart. T-ALL comprises 15% of ALL in pediatric population and 25% of adult cases. T-LBL commonly affects children and adolescents with a high male to female ratio; however, adults can be affected and offer a dismal outcome (17).
Subtypes
Traditionally T-LBL/ALL were classified into L1 to L3 subtypes based upon the nuclear size, nucleocytoplasmic ratio, and nucleoli. However, this classification is now obsolete due to lack of clinical correlation. Currently, the T-LBL/ALL are classified into four subgroups based upon their immunophenotype [European Group for the Immunologic Classification of Leukaemia (EGIL) classification] (20). The common markers which are positive are TdT and cytoplasmic CD3 (CyCD3). The four groups include:
- TI: pro-T-ALL, CD7+ (surface CD3–);
- TII: pre-T-ALL, CD2+ (±CD5, CD8, surface CD3–);
- TIII: cortical T-ALL, CD1a+ (±surface CD3);
- TIV: mature T-ALL, surface CD3+ (CD1a–).
Few studies combine pro- and pre-T-ALL to label as early T-ALL (CD8, surface CD3–, and CD1a–). Many cases from the TI and TII fit into early precursor T-ALL (EPT-ALL) those cases which express myeloid markers like CD13, CD33 with the absence of CD5, and MPO (17,21).
Clinical course
T-LBLs frequently present as mediastinal and nodal masses in the male pediatric population with a second peak in the older population. Bone marrow can get involved during disease but at presentation usually is unaffected or has a minimal blast population. Extranodal sites such as pleuro-pericardium, CNS, skin, and testes can be involved. Mediastinal involvement can present as a rapidly growing bulky anterior mediastinal mass (thymic) and lead to complications of pleural and pericardial effusions, superior vena cava syndrome and, tracheal obstruction. High leukocyte count accompanied with high LDH levels can be seen. Bone marrow involvement can manifest as anemia, thrombocytopenia, and recurrent infections. T-LBLs are usually more aggressive than B-LBLs and present at an advanced stage (22-24).
The patients are staged according to the Ann-Arbor staging or St. Jude Children’s Research Hospital staging. Additional hematological, biochemical, CSF analysis, and radiological extent of disease are necessary for appropriate management and prognostication.
Pathophysiology
LBLs are postulated to originate from precursor cells at various stages of their differentiation. Various risk factors such as viruses, immune dysregulation, toxins, radiation have been studied but no definite correlation could be ascertained (25,26). However, certain cytogenetic and chromosomal aberrations have consistently pointed to a possible causative genetic landscape. Various T-cell receptor rearrangements and aberrant expression of transcription factors have been seen to cause T-LBLs.
Histopathology and cytochemistry
Both T-LBL and ALL have an identical histomorphology consisting of effacement of architecture by the monomorphic population of small to intermediate size cells with scant cytoplasm (blasts), finely dispersed to condensed chromatin and inconspicuous to small nucleoli. Frequent mitosis may be seen in T-LBLs and the surrounding tissue is often infiltrated. The cells show periodic acid-Schiff (PAS) positivity, variable nonspecific esterase (NSE) (multifocal, punctuate in golgi region), Sudan black B positivity, and MPO negativity on cytochemistry. T-LBL can show focal acid phosphatase positivity (17).
Immunophenotype and flowcytometry
For the confirmation of the diagnosis and lineage specification, immunophenotyping is essential. T-LBLs are TdT positive and show positivity for other lineage-specific markers. The earliest T marker to become positive is CD7, however, it is not lineage-specific. CyCD3 is the next to appear, followed by CD2, CD5, and surface CD3. Surface CD3 is the most specific lineage-specific marker (27) (Figure 5). The other immaturity markers which are positive in T-LBL/ALL are CD34, CD1a, and CD99. Initially, the cells are double-negative for CD4 and CD8 after which they become double-positive (pro, pre, T). CD79a, a B cell marker can be noted among some T-LBLs; however, CD3 positivity is definite for T-lineage (28). Aberrant expression of myeloid markers such as CD13 and CD33 can be seen. The common immaturity marker like CD34 which present in acute leukemias is absent in most of the cases of ALL (taking 10% cut off present in 40% cases). LIM domain only 2 (LMO2) expression has been identified as a differentiating marker for T-LBL/ALL. TAL1, a nuclear stain has been useful in some of the cases. Also, ALL and LBL of the same lineage exhibit the same immunophenotype with ALL comprising more of the immature cells (29).

Genetic profile
Among the T-LBL, clonal rearrangements of TCR in all cases along with IgH rearrangement in a fifth of cases have been observed with abnormal karyotype in most of the cases. Alpha and delta loci in chromosome 14, beta in chromosome 7, and gamma in chromosome 7 have been known to cause dysregulation of transcription factors. These rearrangements are associated with a poorer prognosis. BFM Group and French Study reported NOTCH1 and FBXW7 mutation in 50–60% cases followed by loss of heterozygosity (LOH) at 6q. The other notable abnormalities in T-LBL/ALL include phosphatase and tensin homolog (PTEN) deletion, CALM-AF10 fusion, mutations of IL-7R, and CASP8AP2 deletion. Feng et al. described overexpression of Bcl2 and MYC in T-LBL in comparison to ALL. Jeon et al. showed overexpression of promyelocytic leukemia zinc-finger (PLZF) related to bone marrow involvement (30-35). The T-ALL/LBL have also been divided into mutually exclusive genetic subgroups based on the aberrant expression of TAL1/LMO, TLX1/HOX11 which has a favorable prognosis, TLX3/HOX11L2, and HOXA gene having poor prognosis (36).
Differential diagnosis
Based on clinical and radiological findings
The differential diagnosis of pediatric mediastinal mass depends upon the anatomical site and age. Mass in the anterior compartment mainly includes (I) TLBL; (II) HL; (III) PMBCL; (III) germ cell tumor; (IV) primitive neuroectodermal tumor (PNET); (V) thymoma (rare in pediatric age group). Middle mediastinum lesions include mainly vascular anomalies and cysts (bronchogenic cyst, esophageal cyst). Anterior mediastinal lymphomas can also extend to the middle mediastinum (Figure 6). Posterior mediastinum masses include (I) neuroblastoma and its subtypes; (II) nerve sheath tumors (37). The differentials in the spectrum of lymphomas include mainly T-LBL, HL, and PMBCL, all of which have a similar radiological appearance. It is essential to differentiate between the above given optimal patient care and histopathology serves as the bridge. Morphologically, thymoma is the closest differential of T-LBL. Leukemic presentation and bone marrow involvement help in confirmation of T-LBL. As most of the tissues received are needle core biopsies, it is required to differentiate normal thymic tissue from thymoma (B1& B2) and thymic hyperplasia. The age of presentation plays a role as thymomas are unusual in pediatric age group. As shown above, the normal thymus shows the presence of epithelial cells highlighted by cytokeratin stain. Thymoma also shows diffuse cytokeratin (AE1/AE3, Ck5/6) network. However, LBL shows the absence of epithelial meshwork (Figure 7). Immaturity markers (TdT, CD1a, CD99) will be positive in immature lymphocytes of all the cases. In addition to immunohistochemistry (IHC), flowcytometric (FC) analysis will help in differentiation. FC shows a distinct pattern of maturation of thymocytes. There is a continuous maturation pattern showing CyCD3 and surface CD3 in thymoma. Similar pattern is seen in CD4–/CD8– to CD4+ or CD8+ cells. Presence of a clear discrete cluster of cells with dim CD45 and co-expression of CD10 and CD34 favors LBL. LBL also shows aberrant antigen expression like the presence of CD13, CD33 with an absence of T cell marker (CD2 or CD5) (38). LMO2, a transcription factor for hematopoiesis helps to differentiate normal precursor T cells from LBL T cells. LMO2 is weakly positive in thymic epithelial cells while shows strong positivity in B cells and immature T cells of LBL, thereby helps differentiate normal thymus and thymoma where it is negative from LBL which shows strong nuclear positivity (39). The other common differential diagnosis in this age group, the PNET presents radiologically as a mass arising from chest wall infiltrating into anterior mediastinum. CD99 (MIC2) will not help as is positive in both the conditions. PNET cells are immunopositive for FLI1, MIC2 while they are negative for TdT, LCA, and CD3. Neuroblastoma shows similar morphology but it arises in posterior mediastinum and LCA and CD3 negativity rules out T-LBL.


Treatment and prognosis
Detailed treatment is out of the scope of this article. Multidrug chemotherapy has proved to be of benefit to the patient including cyclophosphamide, methotrexate, prednisone, and vincristine. Intrathecal chemotherapy is of great benefit in the intrathecal spread and prevents relapse. With newer drugs targeted against the CD3 receptor, CD52 can help in a better outcome. Since mediastinum is the most frequent site of recurrence and treatment failure, intensive chemotherapy and local radiotherapy have proved to be beneficial. Complications of treatment include febrile neutropenia, tumor lysis syndrome, and infections. For better patient outcomes, rigorous monitoring with radiological, hematological, and FC investigations is necessary. Stem cell transplantation can improve long term survival. B-LBL tends to have a better prognosis than T-LBL.
Poor prognostic factors include adult, female patients presenting with high LDH levels, CNS involvement, and refractory cases. The clinical factors which are responsible for poor outcome include superior mediastinal syndrome (SMS), pleural infusions at initial presentation, SMS with baseline ECOG PS >2, high leucocyte count, low serum albumin, and duration of symptoms (23,24). The genetic abnormalities which are associated with good prognosis include chromosomal rearrangements involving TAL1 [t(1:14)], t(1:17), TALX1, HOXA (CALM-AF10), deletion CDKN2A/2B, mutation in NOTCH1 and those associated with poor prognosis include rearrangements of TLX3 and mutation involving EZH2 (27).
PMBCL
PMBCL is a subtype of large B-cell lymphoma which arises from thymic B cell and has a characteristic gene expression profile in comparison to other large B cell lymphomas (LBCLs) and distinct clinical features. Historically (pre-IHC era) these lymphomas were known as undifferentiated thymic carcinoma and were later coined as histiocytic lymphoma and clear cell lymphoma (10). It comes under the category of “other large B cell lymphoma” as per WHO 2017 classification of hematolymphoid neoplasm (40).
Clinical feature
PMBCL comprises 2–3% of all NHLs (40). It mostly affects the female population with a M:F ratio 1:2. However, Liu et al. reported M:F of 1:1 among the African American population (41). The age of presentation is a young adult with a median age of 35. It can be seen in the adolescent age group. It presents as an anterosuperior mediastinal mass with bulky disease (>10 cm). The clinical manifestation is due to mass effect causing superior vena cava syndrome seen in 25–30% cases as the initial presentation. The common presenting symptom is cough and dyspnea (10,40). The mass usually involves the adjacent organs—lung and pleura and may present as a pleural and pericardial effusion, which seen in 30% of cases.
Extra-mediastinal lymph node involvement is rare; however regional mediastinal node involvement can be seen. Few cases show supraclavicular and cervical lymph node involvement along with the anterior mediastinal mass. Leukemic presentation and bone marrow involvement usually not seen (40). In the advanced stage of the disease, distant organ involvement such as kidney, adrenal, brain, testes can be seen. Most of the patients present in early-stage (I or II) disease. Imaging shows a bulky anterior mediastinal mass with infiltration to adjacent organs. Mediastinal lymphadenopathy may be seen.
Microscopy
Microscopy shows diffuse growth pattern with fibrosis. The fibrotic bands form vague nodules or may show an alveolar pattern. The nodular appearance resembles nodular sclerosis type HL. Focal areas show pericellular fibrosis. The tumor cells usually infiltrate the capsule involving the perithymic fat and fibro collagenous tissue. The cells are intermediate to large-sized with clear cytoplasm, some of which may show eosinophilic cytoplasm. The clear cells and eosinophilic cells are intermixed in most of the cases. Due to fibrosis and the presence of clear cytoplasm; historically these lymphomas were termed sclerotic lymphoma or clear cell lymphoma (Figure 8). The nucleus is vesicular with an irregular nuclear membrane. Multilobate cells resembling Reed-Sternberg (RS) cells may be seen. Unlike HL, there is a paucity of an inflammatory background of plasma cells and eosinophils. Areas of necrosis may be seen. Due to marked fibrosis, in some case needle core biopsies interpretation may be difficult and show only marked crushing artifact (10,40).

IHC and molecular profile
As the cell of origin is the thymic B-cell, PMBCL shows strong and diffuse immunopositive for all B cell markers CD20, CD19, CD79a, and PAX5. It also shows strong positivity for LCA. The B-cell transcription factors—OCT2 and BOB1 are retained. The positivity of other markers helpful in diagnosis is CD30 (weak and variable), CD23 (80% cases), CD200, MAL, TRAF, CREL, and PDL1 (8,42-45). As described earlier, thymic B-cells resemble post germinal center B-cells, hence most (75%) of PMBCL are MUM1 positive, and in few cases show CD10 and BCL6 positivity. CD15 is usually absent, however, an occasional case reported CD15 positivity (Figure 9). The cells are consistently negative for EBER-ISH and EBV-LMP (40).

PMBCL usually lacks immunoglobulin rearrangement which helps differentiate from other B cell lymphomas. The gold standard for diagnosis is the gene expression profile analysis which can diagnose PMBCL in 80% cases. Recently, Mottok et al. described a 58 gene panel that can be done with RNA extracted from formalin fix paraffin-embedded tissue based on NanoString technology. It can differentiate PMBCL and DLBCL with 92.9% accuracy (46,47). Other notable genomic profile includes amplification 9p24 (PDL1/PDL2) loci.
Though PMBCL resembles LBCL immunohistochemically and morphologically; however, the genetic profile resembles HL. There is an amplification of 9p24 which encodes for PDL1/PDL2 loci, CIITA alteration, and JAK2 alteration which also seen in HL (40).
Differential diagnosis
Radiologically, the differentials remain the same as discussed above in the T-LBL section. In pediatric age group, PMBCL is rare in comparison to LBL and HL. Histomorphology helps in differentiating immature lymphomas like LBL from lymphomas of mature origin like HL and PMBCL. Large cells with vesicular nuclei and abundant cytoplasm and the presence of fibrosis favors HL/PMBCL/DLBCL NOS. Diffuse CD20 positivity favors PMBCL or LBCL of non-thymic origin over HL. However, 10% of HLs can show diffuse CD20 positivity. However, CD20 HL is weak and variable with dim PAX5. HL shows inflammatory background (eosinophil, plasma cells, and mature lymphocytes) which is absent in PMBCL. The RS cells are immunopositive for CD30 (strong), CD15 while they are negative for LCA. The background inflammatory cells comprise of CD3 positive small lymphocytes which are absent very sparse in PMBCL. Aladily et al. described a combination of three markers—CD83+, fascin+, and CD23– (HL) and CD83–, fascin–, and CD23+ (PMBCL) which is 100% specific in differentiating HL and PMBCL (Table 1) (48). As shown above, all the mediastinal compartments contain multiple lymph nodes. LBCLs can arise from lymph nodes other than Thymic B cell. The treatment modalities are different for LBCL [diffuse LBCL (DLCBL), NOS] of non-thymic origin and PMBCL, thereby necessitating their differentiation. The helpful markers include CD23, CD200, CREL, MAL which are positive in PMBCL in comparison to LBCL, non-thymic type. The most useful marker is the absence of immunoglobulin rearrangement which can be demonstrated by in-situ hybridization for Kappa and lambda negativity (Table 2).
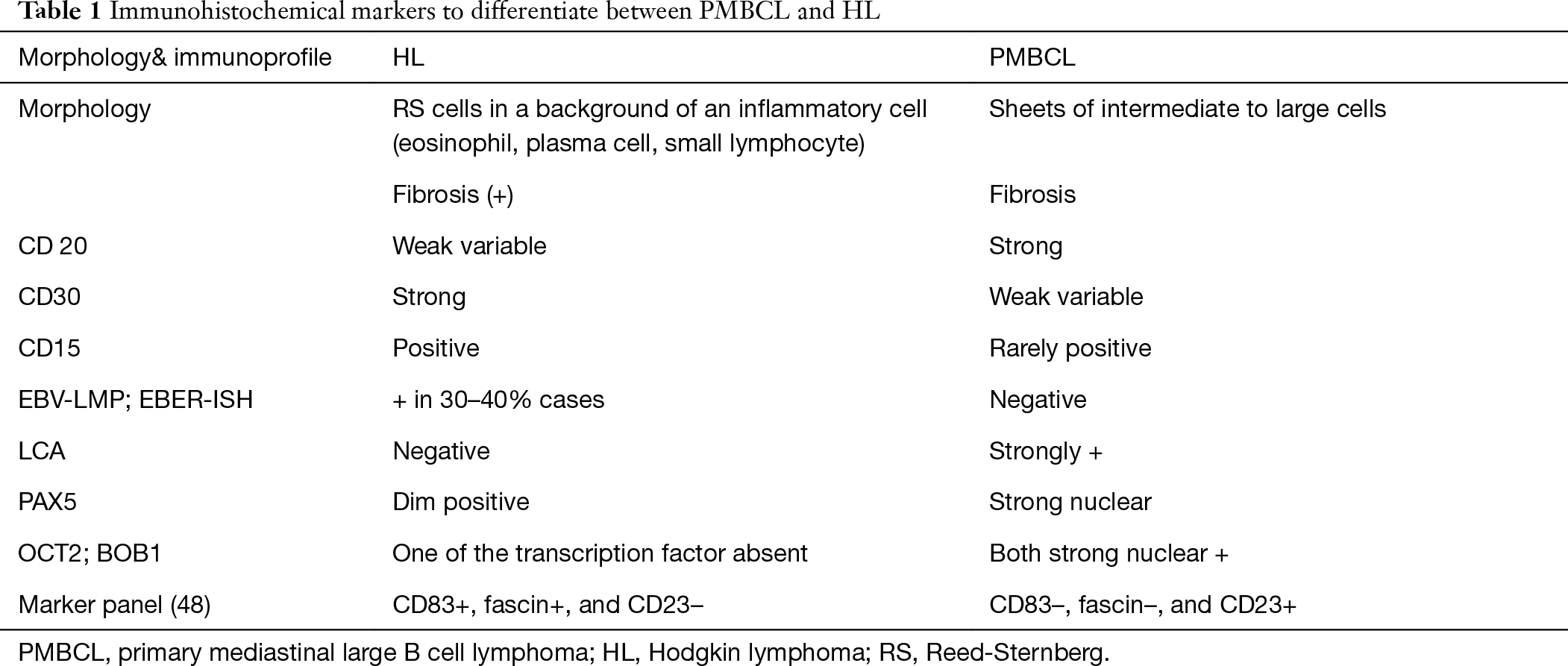
Full table

Full table
Among the T-cell lymphomas, ALK-positive anaplastic large cell lymphoma could be a possibility morphologically, however LCA and CD20 diffuse positivity rules out ALCL. The other close mimickers in pediatric age group are germ cell tumors which are LCA negative and SALL4 positive.
Treatment and prognosis
PMBCL responds well to chemo (DA-EPOCH) and radiotherapy. Prognosis is favorable in comparison to other LBCLs and grey zone lymphomas. Despite good response rates, 10–20% of PMBCL relapse or are refractory to standard therapy (49,50). Poor prognostic factors include an extension to other adjacent organs like lung, pleura, pericardium, high IPI, Ki-67 proliferative index ≥70%, stage III, and stage IV disease (51).
HL
Introduction
HLs are the lymphomas characterized by clonal proliferation of germinal center B-cell in the form of Hodgkin/Reed-Sternberg (HRS) cells in an inflammatory milieu. HL primarily affects the lymph nodes; however, involvement of other sites has been noted (52). Among the extranodal sites, the mediastinum is most commonly involved. Hodgkin disease can affect the mediastinum as a primary disease or as part of systemic involvement. At the initial presentation, more than 80% of patients have intrathoracic involvement more than NHLs (53). Anterior mediastinum and paratracheal nodes are commonly affected in the thorax with spread to contiguous lymph nodes (54). Based upon immunophenotype, clinical feature, the morphology of neoplastic HRS cell, HL broadly classified classical HL and nodular lymphocyte-predominant HL (NLPHL). Classical HL is further subclassified into four subtypes- mixed cellularity, nodular sclerosis, lymphocyte rich, and lymphocyte depleted. Though the immunophenotype of HRS cell remains the same in all the subtypes of classical HL, the epidemiology, inflammatory milieu, prognosis, and histomorphology varies among the different subtypes. The mediastinum is the primary site of involvement is seen mostly in classical HL (mainly nodular sclerosis type); however, is a rare occurrence is seen in NLPHL (52,54). This review will focus more on the Nodular sclerosis subtype.
Incidence
Lymphomas form the major bulk of anterior mediastinal masses in children (37). Though HLs comprise less than 1% of the newly diagnosed cancer cases worldwide with a prevalence of 2–4 cases per 100,000 populations, it constitutes approximately 50% of mediastinal lymphomas (54-56). Thymic enlargement is found in 28–30% of pediatric population with HL. In contrast to PMBCL where thymus is the predominant site of disease, only thymic involvement in HL is rare (2.8%) (57). Of the HLs irrespective site of presentation, 90% are the classical, and rest represent NLPHL. Among the classical HL 70% are nodular sclerosis and 20–25% mixed cellularity. However, in mediastinum 95% comprises nodular sclerosis (NS-CHL) and 5% mixed cellularity MC-CHL (52,54).
Clinical course
Though immunophenotypically and genetically all the classical HL share common features, the clinical features vary among subtypes. NS-CHL is more common among the urban population with good socioeconomic status in comparison to MC-CHL which is common in developing countries. NS-CHL present in adolescents and young adults with a peak between 15–34 years and has female predominance. MC-CHL has a bimodal age distribution with adults (40–50 years) and children being affected the most. With a predominant nodal involvement, patients present with peripheral lymphadenopathy and B-symptoms. Cervical lymphadenopathy is the most common site followed by abdominal lymph nodes. Nonaxial nodes like mesenteric and epitrochlear nodes are usually spared. Patients present usually with longer duration of symptoms in comparison to aggressive lymphomas. Most cases of mediastinal HL are asymptomatic and incidentally detected. Patients with large mass present with symptoms of dyspnea, dysphagia, retrosternal chest pain, and cough. Bulky masses can cause SVC syndrome and effusions but less in comparison to aggressive lymphomas. Patients with deferred immunity as on immunosuppression (HIV) or with autoimmune diseases are prone to develop primary mediastinal HL (3). An elevated LDH and ESR levels correspond to the bulky disease. Radiological findings suggest nodal and visceral involvement. Patients staged according to the Ann Arbor staging. Bone marrow involvement upstages the disease to stage IV. An international prognostic index can be used for prognostication (52,54,58).
Histopathology
In the current era of practice, as reiterated above most of the mediastinal biopsies are needle core with no role of gross morphology. The excisional biopsies, if taken from peripheral lymph node show enlargement and are matted with a rubbery consistency. Cut surface shows multiple nodules surrounded by fibrous septa. Cystic changes may be seen in the thymus. MC-CHL lymph nodes show homogeneous pale yellow appearance. On microscopy, the nodes have effaced architecture and show the presence of RS cells which are large cells with abundant amphophilic to basophilic cytoplasm, prominent eosinophilic nucleoli, and perinucleolar clearing. The background consists of a mixed inflammatory cell population and is rich in eosinophils (Figure 10A,B). Fibrosis and the inflammatory milieu are the results of excessive cytokines release by the RS cells. Nodular sclerosis subtype is characterized by a nodular growth pattern surrounded by fibroblast poor collagen septa. The RS cells of NS-CHL show more nuclear location with small nucleoli, more abundant cytoplasm, and cytoplasmic retraction, giving the name of lacunar type RS cells. Areas of necrosis with an aggregation of histiocytes, mimicking a granuloma can be seen (52,54,58). Thymic involvement can particularly lead to cystic changes. Tumor burden, degree of sclerosis, and amount with atypical of RS cells are of surmounting importance; and has been correlated with prognosis (59). Based upon the presence of large pleomorphic cells, British National Lymphoma Investigation subtyped NS-CHL into two categories—BNLI grade 1 and grade 2. Grade 2 (higher grade) is defined by the presence of >25% nodule showing pleomorphic HRS cell without lymphocyte depletion or >25% of the cellular nodules showing pleomorphic or reticular lymphocyte depletion or >80% showing fibrohistiocytic variant. Mixed cellularity type shows a diffuse pattern of involvement with classical RS cells (binucleate cells prominent large eosinophilic nucleoli with perinuclear halo). The inflammatory milieu may predominate by any one type of cell. Lymphocyte depleted and lymphocyte rich subtypes constitute <5% of total HL subtypes and extremely rare in the mediastinum, not discussed in this review (52).

Immunophenotype
The HRS cells of classical HL show membranous with golgi zone accentuation for CD30 (>90%) and CD15 (60–70%). Despite the cell of origin of HL from B cell, immunophenotypically B cell antigens are not fully expressed in HL. The HRS cells are negative or variable positive for CD20 (weak positive in 20–30%) and CD79a (<10% cases), however, PAX5 shows dim positivity in almost all cases. MUM1 positivity is seen in 90% of HRS cells (60) (Figure 10C,D,E,F). The B cell transcription factors, OCT2 and BOB1 can be negative or show positivity of anyone of the markers. CD45 and EMA are negative. In mixed cellularity type, the EBV association can be demonstrated by positivity for EBV-LMP and EBER-ISH positivity in 60% of cases. EBER is more sensitive than EBV-LMP (61). Few of the cases show aberrant or artefactual expression of T-cell markers (CD2 and CD7) in the RS cells (62).
NLPHL type characteristically retains expression of B-cell markers—CD20, CD79a, PAX5, OCT2, BOB1, and CD45. The RS cells are CD30 and CD15 negative and may show variable positivity for immunoglobulin chains. The follicular dendritic meshwork is highlighted by CD21/CD23. T-cell rosettes highlighted by PD1 positive T-follicular helper T-cells can aid the diagnosis (52).
Genetic profile
The neoplastic cells are derived from the germinal center B-cells and rearrangements in the immunoglobulin gene are common. A plausible role of Epstein Barr virus has been established in HL (particularly mixed cellularity and lymphocyte depleted type). EBV (LMP protein) infection upregulates the NF-kB pathway thereby leading to increased proliferation and decreased apoptosis of B-cells. Mutation in the immunoglobulin gene is acquired which causes the production of RS cell. A histopathological examination is required to differentiate it from infectious and other neoplastic causes (52,54).
HL has a varied mutational landscape and genetic profile. Primarily a disease of B-cells, mutations in signaling pathways like JAK/STAT, NF-kB, GM-CSF/IL-3, CBP/EP300 along with gene variants affecting the B-cell receptor (BCR) pathway, such as BTK, CARD11 have been detected (63). Clonal rearrangements of immunoglobulin heavy and light chains have been detected in both classical and NLPHL, thereby supporting their derivation from B-cell lineage. However, rarely rearrangements in T-cell receptor are also noted (64,65). Mutations in JAK/STAT regulation leading to blocked negative feedback causes nuclear accumulation of STAT5 like mediastinal large cell lymphoma (66,67). Overexpression of p53 protein is noted; however, mutation of TP53 is rare (68). PD1 ligand overexpression is also noted (69). Loss of MHC I is also noted (70). Gains of chromosomal arms of 2p, 12q, 9p, and loss of 6q, 13 q are seen. Aneuploidy and hypertetraploidy observed corroborate to the multinucleation (52).
Differential diagnosis
Clinically and radiologically the differential diagnosis is the same as discussed in PMBCL sections. In mediastinum, composite lymphomas (both HL and PMBCL) and Grey zone lymphoma (lymphoma with features intermediate between HL and PMBCL) is more common than lymphoma at other anatomical sites. Due to needle core nature of biopsies, regional variability is difficult to appreciate. Evidence of the sequential presence of HL followed by PMBCL and vise-versa has also been noted.
Treatment and prognosis
HL with the current therapeutics has a maximal 5-year survival with cure achieved in most of the patients. Polychemotherapy regimen including doxorubicin, bleomycin, vinblastine, and dacarbazine (ABVD regimen) and BEACOPP including bleomycin, etoposide, doxorubicin, cyclophosphamide, vincristine procarbazine and prednisolone along with radiotherapy have a great cure rate. However, higher stage, mediastinal involvement, high ESR, more than three groups of nodal involvement confers a higher risk of relapse and refractory disease. For the refractory cases, targeted therapy like brentuximab for CD30 is also available. PET scan for timely evaluation of response ensures early detection of relapse and better patient outcome.
Conclusions
Pediatric mediastinal lymphomas constitute significant proportion of pediatric mediastinal tumors. Clinically and radiologically the major mediastinal lymphomas TLBL, HL and PMBCL mimics closely. In the current era due to limited availability of tissue (core needle biopsy) care of handling of tissue sample is required for optimal utilization. Multidisciplinary approach including extended panel IHC, in-situ hybridization, flowcytometry and molecular profiling is required for proper diagnosis and treatment.
Acknowledgments
We would like to thank Dr. Jagdish P. Meena for help with the radiological image and Dr. Deepam Pushpam for help with Figure 4.
Funding: None.
Footnote
Provenance and Peer Review: This article was commissioned by the Guest Editor (Deepali Jain) for the series “Pediatric Mediastinal Tumors” published in Mediastinum. The article was sent for external peer review organized by the Guest Editor and the editorial office.
Reporting Checklist: The authors have completed the Narrative Review checklist. Available at http://dx.doi.org/10.21037/med-20-37
Conflicts of Interest: All authors have completed the ICMJE uniform disclosure form (available at http://dx.doi.org/10.21037/med-20-37). The series “Pediatric Mediastinal Tumors” was commissioned by the editorial office without any funding or sponsorship. The authors have no other conflicts of interest to declare.
Ethical Statement: The authors are accountable for all aspects of the work in ensuring that questions related to the accuracy or integrity of any part of the work are appropriately investigated and resolved.
Open Access Statement: This is an Open Access article distributed in accordance with the Creative Commons Attribution-NonCommercial-NoDerivs 4.0 International License (CC BY-NC-ND 4.0), which permits the non-commercial replication and distribution of the article with the strict proviso that no changes or edits are made and the original work is properly cited (including links to both the formal publication through the relevant DOI and the license). See: https://creativecommons.org/licenses/by-nc-nd/4.0/.
References
- Burlew JT, Banks KP. Anatomy, thorax, mediastinal lymph nodes. In: StatPearls. Treasure Island: Stat Pearls Publishing, 2020.
- Pearse G. Normal structure, function and histology of the thymus. Toxicol Pathol 2006;34:504-14. [Crossref] [PubMed]
- De Waal EJ, Schuurman HJ, Van Loveren H, et al. Differential effects of 2,3,7,8-tetrachlorodibenzo-p-dioxin, bis(tri-n-butyltin) oxide and cyclosporine on thymus histophysiology. Crit Rev Toxicol 1997;27:381-430. [Crossref] [PubMed]
- Hofmann WJ, Momburg F, Moller P. Thymic medullary cells expressing B lymphocyte antigens. Hum Pathol 1988;19:1280-7. [Crossref] [PubMed]
- Perera J, Huang H. The development and function of thymic B cells. Cell Mol Life Sci 2015;72:2657-63. [Crossref] [PubMed]
- Nango K, Inaba M, Inaba K, et al. Ontogeny of thymic B cells in normal mice. Cell Immunol 1991;133:109-15. [Crossref] [PubMed]
- Wilson A, MacDonald HR, Radtke F. Notch 1-deficient common lymphoid precursors adopt a B cell fate in the thymus. J Exp Med 2001;194:1003-12. [Crossref] [PubMed]
- Calaminici M, Piper K, Lee AM, et al. CD23 expression in mediastinal large B-cell lymphomas. Histopathology 2004;45:619-24. [Crossref] [PubMed]
- Fend F, Nachbaur D, Oberwasserlechner F, et al. Phenotype and topography of human thymic B cells. An immunohistologic study. Virchows Arch B Cell Pathol Incl Mol Pathol 1991;60:381-8. [Crossref] [PubMed]
- Piña-Oviedo S, Moran CA. Primary mediastinal nodal and extranodal non-Hodgkin lymphomas: current concepts, historical evolution, and useful diagnostic approach: Part 1. Adv Anat Pathol 2019;26:346-70. [Crossref] [PubMed]
- Takeda S, Miyoshi S, Akashi A, et al. Clinical spectrum of primary mediastinal tumors: a comparison of adult and pediatric populations at a single Japanese institution. J Surg Oncol 2003;83:24-30. [Crossref] [PubMed]
- Simpson I, Campbell PE. Mediastinal masses in childhood: a review from a pediatric pathologist’s point of view. Prog Pediatr Surg 1991;27:92-126. [Crossref] [PubMed]
- Tansel T, Onursal E, Dayloğlu E, et al. Childhood mediastinal masses in infants and children. Turk J Pediatr 2006;48:8-12. [PubMed]
- Cortelazzo S, Ferreri A, Hoelzer D, et al. Lymphoblastic lymphoma. Crit Rev Oncol Hematol 2017;113:304-17. [Crossref] [PubMed]
- Giulino-Roth L. How I treat primary mediastinal B-cell lymphoma. Blood 2018;132:782-90. [Crossref] [PubMed]
- Perwein T, Lackner H, Ebetsberger-Dachs G, et al. Management of children and adolescents with gray zone lymphoma: a case series. Pediatr Blood Cancer 2020;67:e28206 [Crossref] [PubMed]
- Borowitz MJ, Chan JKC. T lymphoblastic leukaemia/lymphoma. In: Swerdlow SH, Campo E, Harris NL, et al. editors. World Health Organization classification of tumors of haematopoietic and lymphoid tissues. 4th ed. Lyon: IARC, 2008:176-8.
- Clavel J, Goubin A, Auclerc MF, et al. Incidence of childhood leukaemia and non-Hodgkin's lymphoma in France: National Registry of Childhood Leukaemia and Lymphoma, 1990-1999. Eur J Cancer Prev 2004;13:97-103. [Crossref] [PubMed]
- Han X, Kilfoy B, Zheng T, et al. Lymphoma survival patterns by WHO subtype in the United States, 1973-2003. Cancer Causes Control 2008;19:841-58. [Crossref] [PubMed]
- Bene MC, Castoldi G, Knapp W, et al. Proposals for the immunological classification of acute leukemias. European Group for the Immunological Characterization of Leukemias (EGIL). Leukemia 1995;9:1783-6. [PubMed]
- Coustan-Smith E, Mullighan CG, Onciu M, et al. Early T-cell precursor leukaemia: a subtype of very high-risk acute lymphoblastic leukaemia. Lancet Oncol 2009;10:147-56. [Crossref] [PubMed]
- Kapur S, Levin MB. Precursor T-cell lymphoblastic lymphoma presenting as cardiac tamponade in a 25-year-old male: a case report and review of literature. World J Oncol 2014;5:129-34. [PubMed]
- Patel A, Tiwari A, Biswas B, et al. Clinical predictors and prognostic model for pediatric lymphoblastic lymphoma treated with uniform BFM90 protocol: a single-center experience of 65 patients from Asia. Clin Lymphoma Myeloma Leuk 2019;19:e291-8. [Crossref] [PubMed]
- Tilak TV, Raina V, Kumar L, et al. Superior vena cava syndrome and poor performance status at presentation affect survival in mediastinal T-lymphoblastic lymphoma--a single institute experience from India. Ann Hematol 2013;92:917-23. [Crossref] [PubMed]
- Goedert JJ, Cote TR, Virgo P, et al. Spectrum of AIDS-associated malignant disorders. Lancet 1998;351:1833-9. [Crossref] [PubMed]
- Engels EA, Cerhan JR, Linet MS, et al. Immune-related conditions and immune-modulating medications as risk factors for non-Hodgkin’s lymphoma: a case-control study. Am J Epidemiol 2005;162:1153-61. [Crossref] [PubMed]
- You MJ, Medeiros J, Hsi ED. T-lymphoblastic leukaemia/lymphoma. Am J Clin Pathol 2015;144:411-22. [Crossref] [PubMed]
- Pilozzi E, Muller-Hermelink HK, Falini B, et al. Gene rearrangements in T-cell lymphoblastic lymphoma. J Pathol 1999;188:267-70. [Crossref] [PubMed]
- Weiss LM, Bindl JM, Picozzi VJ, et al. Lymphoblastic lymphoma: an immunophenotype study of 26 cases with comparison to T cell acute lymphoblastic leukemia. Blood 1986;67:474-8. [Crossref] [PubMed]
- Aster JC, Blacklow SC, Pear WS. Notch signalling in T cell lymphoblastic leukaemia/lymphoma and other haematological malignancies. J Pathol 2011;223:262-73. [Crossref] [PubMed]
- Bonn BR, Rohde M, Zimmermann M, et al. Incidence and prognostic relevance of genetic variations in T-cell lymphoblastic lymphoma in childhood and adolescence. Blood 2013;121:3153-60. [Crossref] [PubMed]
- Callens C, Baleydier F, Lengline E, et al. Clinical impact of NOTCH1 and/or FBXW7 mutations, FLASH deletion, and TCR status in pediatric T-cell lymphoblastic lymphoma. J Clin Oncol 2012;30:1966-73. [Crossref] [PubMed]
- Yu H, Du Y, Xu J, et al. Prognostic relevance of genetic variations in T-cell acute lymphoblastic leukemia/lymphoblastic lymphoma. Transl Cancer Res 2019;8:2485-95. [Crossref]
- Feng H, Stachura DL, White RM, et al. T-lymphoblastic lymphoma cells express high levels of BCL2, S1P1, and ICAM1, leading to a blockade of tumor cell intravasation. Cancer Cell 2010;18:353-66. [Crossref] [PubMed]
- Jeon YK, Go H, Nam SJ, et al. Expression of the promyelocytic leukemia zinc-finger in T-lymphoblastic lymphoma and leukemia has strong implications for their cellular origin and greater association with initial bone marrow involvement. Mod Pathol 2012;25:1236-45. [Crossref] [PubMed]
- Meijerink JP. Genetic rearrangements in relation to immunophenotype and outcome in T-cell acute lymphoblastic leukaemia. Best Pract Res Clin Haematol 2010;23:307-18. [Crossref] [PubMed]
- Ranganath SH, Lee EY, Restrepo R, et al. Mediastinal masses in children. AJR Am J Roentgenol 2012;198:W197-216 [Crossref] [PubMed]
- Boddu P, Thakral B, Alhuraiji A, et al. Distinguishing thymoma from T-lymphoblastic leukaemia/lymphoma: a case-based evaluation. J Clin Pathol 2019;72:251-7. [Crossref] [PubMed]
- Jevremovic D, Roden AC, Ketterling RP, et al. LMO2 is a specific marker of T-lymphoblastic leukemia/lymphoma. Am J Clin Pathol 2016;145:180-90. [Crossref] [PubMed]
- Gaulard P, Harris NL, Pileris SA, et al. Primary mediastinal (thymic) large 8-cell lymphoma. In: Swerdlow SH, Campo E, Harris NL, et al. editors. WHO classification of tumours of haematopoietic and lymphoid tissues (revised 4th edition). Lyon: IARC, 2017:314-18.
- Liu PP, Wang KF, Xia Y, et al. Racial patterns of patients with primary mediastinal large B-cell lymphoma: SEER analysis. Medicine (Baltimore) 2016;95:e4054 [Crossref] [PubMed]
- Copie-Bergman C, Plonquet A, Alonso MA, et al. MAL expression in lymphoid cells: further evidence for MAL as a distinct molecular marker of primary mediastinal large B-cell lymphomas. Mod Pathol 2002;15:1172-80. [Crossref] [PubMed]
- Dorfman DM, Shahsafaei A, Alonso MA. Utility of CD200 immunostaining in the diagnosis of primary mediastinal large B cell lymphoma: comparison with MAL, CD23, and other markers. Mod Pathol 2012;25:1637-43. [Crossref] [PubMed]
- Rodig SJ, Savage KJ, LaCasce AS, et al. Expression of TRAF1 and nuclear c-Rel distinguishes primary mediastinal large cell lymphoma from other types of diffuse large B-cell lymphoma. Am J Surg Pathol 2007;31:106-12. [Crossref] [PubMed]
- Guiter C, Dusanter-Fourt I, Copie-Bergman C, et al. Constitutive STAT6 activation in primary mediastinal large B-cell lymphoma. Blood 2004;104:543-49. [Crossref] [PubMed]
- Scott D, Wright G, Williams M, et al. Accurate diagnosis of aggressive B cell non-Hodgkin lymphomas using gene expression profiling of formalin-fixed, paraffin-embedded tissues. Blood 2014;124:3016. [Crossref]
- Mottok A, Wright G, Rosenwald A, et al. Molecular classification of primary mediastinal large B-cell lymphoma using routinely available tissue specimens. Blood 2018;132:2401-5. [Crossref] [PubMed]
- Aladily TN, Mansour A, Alsughayer A, et al. The utility of CD83, fascin and CD23 in the differential diagnosis of primary mediastinal large B-cell lymphoma versus classic Hodgkin lymphoma. Ann Diagn Pathol 2019;40:72-6. [Crossref] [PubMed]
- Lees C, Keane C, Gandhi MK, et al. Biology and therapy of primary mediastinal B-cell lymphoma: current status and future directions. Br J Haematol 2019;185:25-41. [Crossref] [PubMed]
- Gogia A, Kumar S, Chellapurum SK, et al. Primary mediastinal B cell lymphoma: a limited institutional experience with uniform DAEPOCH-R protocol. Indian J Hematol Blood Transfusn 2020. doi:
10.1007/s12288-020-01301-z . - Zhou H, Xu-Monette ZY, Xiao L, et al. Prognostic factors, therapeutic approaches, and distinct immunobiologic features in patients with primary mediastinal large B-cell lymphoma on long-term follow-up. Blood Cancer J 2020;10:49. [Crossref] [PubMed]
- Stein H, Pileri SA, Weiss LM, et al. Hodgkin Lymphoma. In: Swerdlow SH, Campo E, Harris NL, et al. editors. WHO classification of tumours of haematopoietic and lymphoid tissues (revised 4th edition). Lyon: IARC, 2017:423-30.
- Castellino RA, Blank N, Hoppe RT, et al. Hodgkin disease: contributions of chest CT in the initial staging evaluation. Radiology 1986;160:603-5. [Crossref] [PubMed]
- Piña-Oviedo S, Moran CA. Primary mediastinal classical hodgkin lymphoma. Adv Anat Pathol 2016;23:285-309. [Crossref] [PubMed]
- Cartwright R, Brincker H, Carli PM, et al. The rise in incidence of lymphomas in Europe. Eur J Cancer 1999;35:627-33. [Crossref] [PubMed]
- Duwe BV, Sterman DH, Musani AI. Tumors of the mediastinum. Chest 2005;128:2893-909. [Crossref] [PubMed]
- Luker GD, Siegel MJ. Mediastinal Hodgkin disease in children: response to therapy. Radiology 1993;189:737-40. [Crossref] [PubMed]
- Wang HW, Balakrishna JP, Pittaluga S, et al. Diagnosis of Hodgkin lymphoma in the modern era. Br J Haematol 2019;184:45-59. [Crossref] [PubMed]
- Bennett MH, MacLennan KA, Easterling MJ, et al. The prognostic significance of cellular subtypes in nodular sclerosing Hodgkin's disease: an analysis of 271 non-laparotomised cases (BNLI report no. 22). Clin Radiol 1983;34:497-501. [Crossref] [PubMed]
- Carbone A, Gloghini A, Aldinucci D, et al. Expression pattern of MUM1/IRF4 in the spectrum of pathology of Hodgkin's disease. Br J Haematol 2002;117:366-72. [Crossref] [PubMed]
- Delsol G, Brousset P, Chittal S, et al. Correlation of the expression of Epstein-Barr virus latent membrane protein and in situ hybridization with biotinylated BamHl-W probes in Hodgkin's disease. Am J Pathol 1992;140:247-53. [PubMed]
- Tzankov A, Bourgau C, Kaiser A, et al. Rare expression of T-cell markers in classical Hodgkin's lymphoma. Mod Pathol 2005;18:1542-9. [Crossref] [PubMed]
- Mata E, Díaz-López A, Martín-Moreno AM, et al. Analysis of the mutational landscape of classic Hodgkin lymphoma identifies disease heterogeneity and potential therapeutic targets. Oncotarget 2017;8:111386-95. [Crossref] [PubMed]
- Deng F, Lü G, Li G, et al. Hodgkin's disease: immunoglobulin heavy and light chain gene rearrangements revealed in single Hodgkin/Reed-Sternberg cells. Mol Pathol 1999;52:37-41. [Crossref] [PubMed]
- Aguilera NS, Chen J, Bijwaard KE, et al. Gene rearrangement and comparative genomic hybridization studies of classic Hodgkin lymphoma expressing T-cell antigens. Arch Pathol Lab Med 2006;130:1772-9. [PubMed]
- Tiacci E, Ladewig E, Schiavoni G, et al. Pervasive mutations of JAK-STAT pathway genes in classical Hodgkin lymphoma. Blood 2018;131:2454-65. [Crossref] [PubMed]
- Savage KJ, Monti S, Kutok JL, et al. The molecular signature of mediastinal large B-cell lymphoma differs from that of other diffuse large B-cell lymphomas and shares features with classical Hodgkin lymphoma. Blood 2003;102:3871-9. [Crossref] [PubMed]
- Montesinos-Rongen M, Roers A, Küppers R, et al. Mutation of the p53 gene is not a typical feature of Hodgkin and Reed-Sternberg cells in Hodgkin's disease. Blood 1999;94:1755-60. [Crossref] [PubMed]
- Green MR, Monti S, Rodig SJ, et al. Integrative analysis reveals selective 9p24.1 amplification, increased PD-1 ligand expression, and further induction via JAK2 in nodular sclerosing Hodgkin lymphoma and primary mediastinal large B-cell lymphoma. Blood 2010;116:3268-77. [Crossref] [PubMed]
- Nijland M, Veenstra RN, Visser L, et al. HLA dependent immune escape mechanisms in B-cell lymphomas: Implications for immune checkpoint inhibitor therapy? Oncoimmunology 2017;6:e1295202 [Crossref] [PubMed]
Cite this article as: Mallick S, Jain S, Ramteke P. Pediatric mediastinal lymphoma. Mediastinum 2020;4:22.

